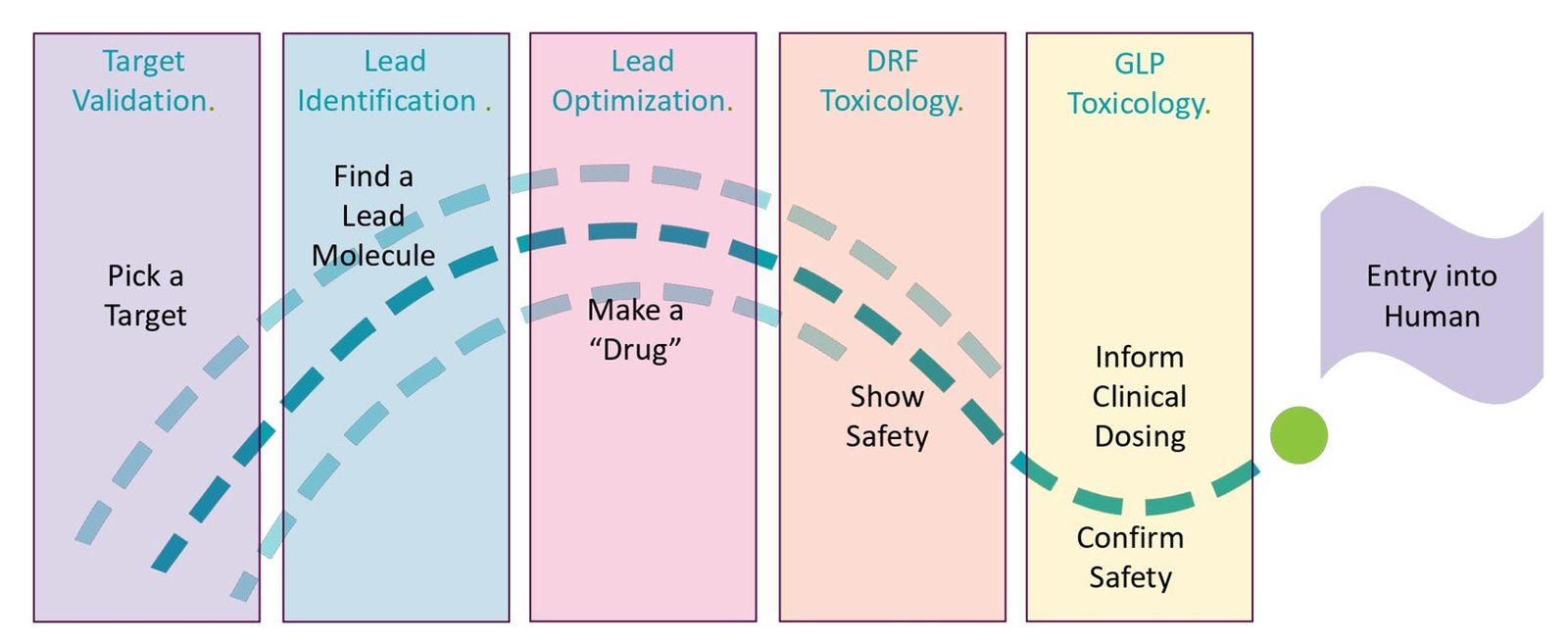
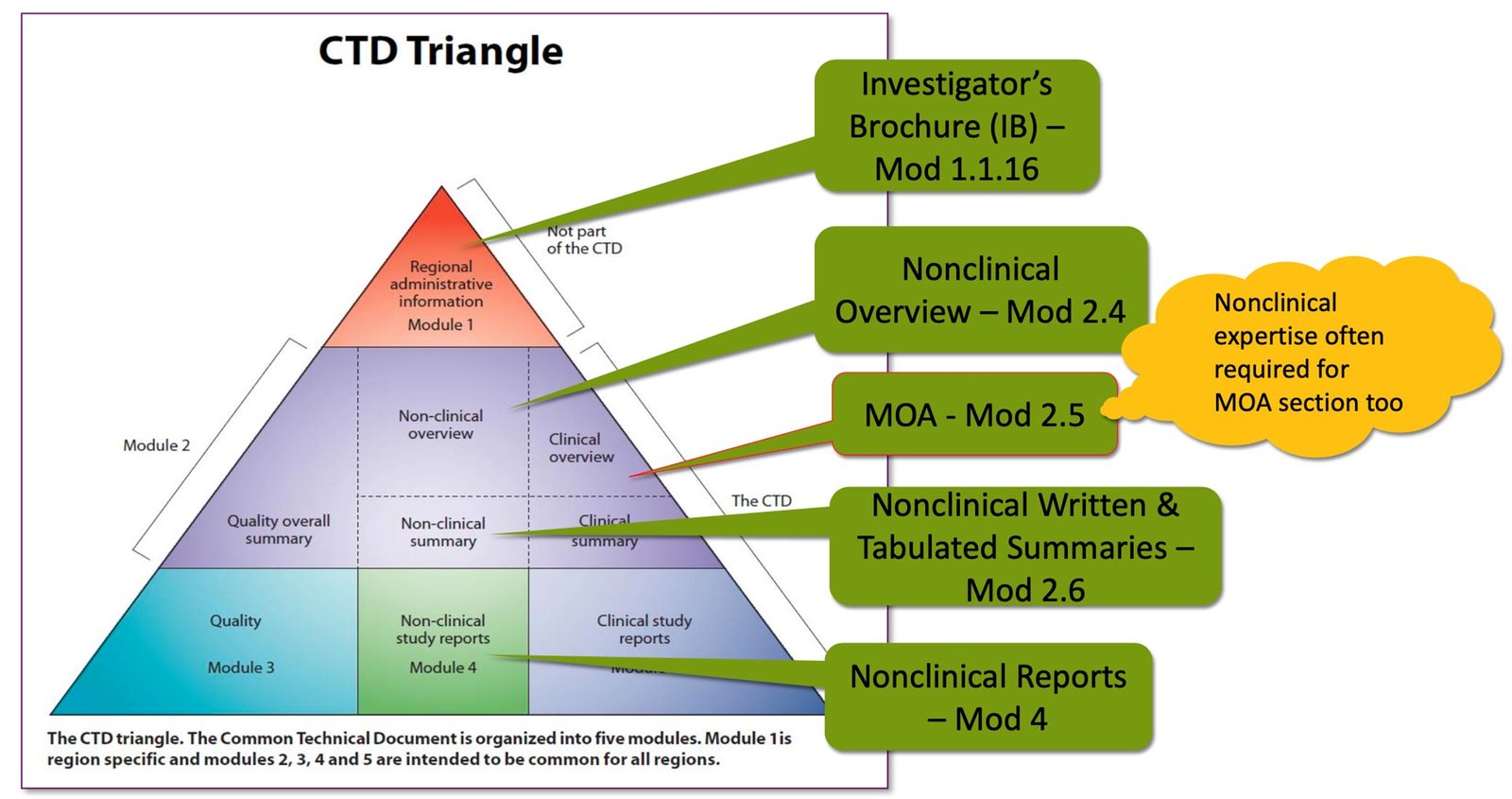
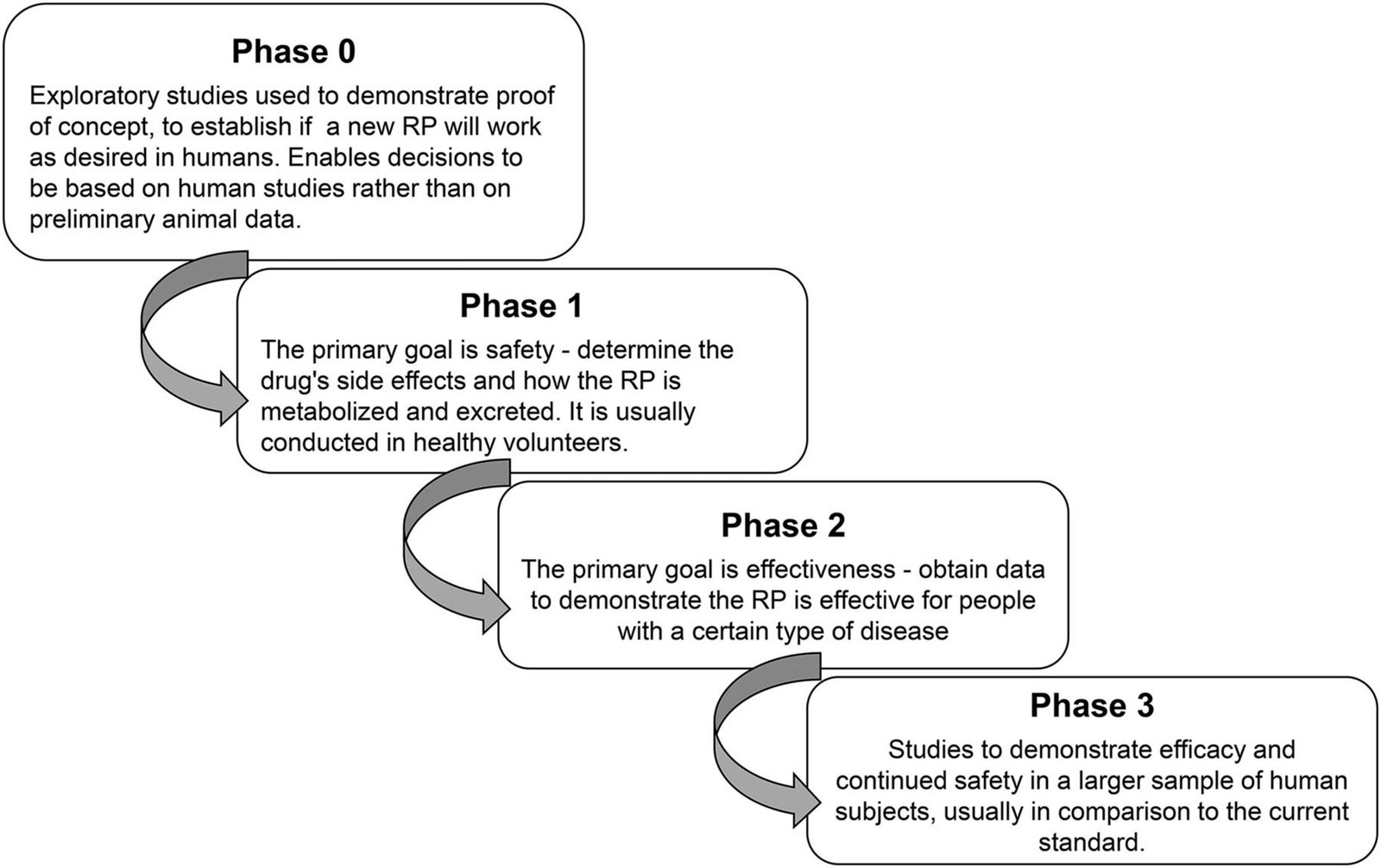
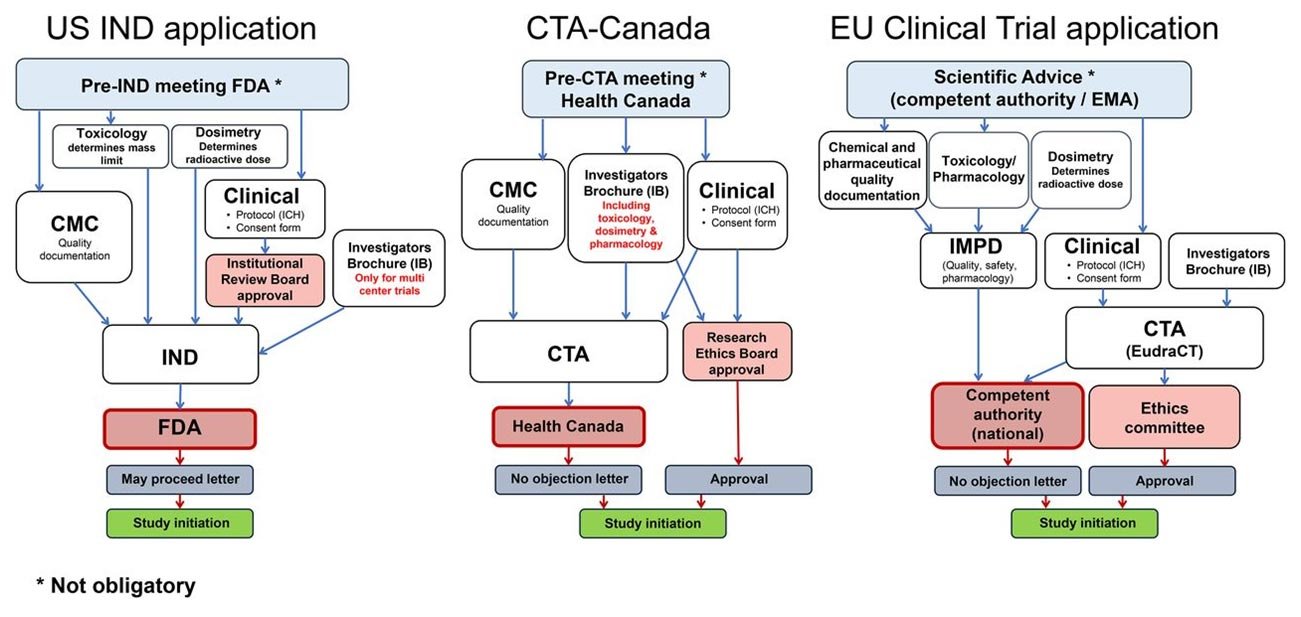
Regulatory strategy development: Creating a comprehensive plan for how a product will navigate regulatory requirements and identifying the most efficient approval pathway. This begins during early product development to minimize delays.
Submission preparation and management: Handling the extensive documentation required for applications to regulatory agencies. This includes:
- Investigational New Drug (IND) applications to begin human clinical trials.
- New Drug Applications (NDA) for approval to market a new drug.
- Biologics License Applications (BLA) for biologic therapies.
- Agency interactions: Preparing for and managing communication with regulatory bodies, including strategy for meetings and responses to information requests.
- Post-market compliance: Assisting with ongoing regulatory obligations after a product is approved, such as required reporting, labeling updates, and safety surveillance.
- Quality management system (QMS) implementation: Helping companies establish or improve the systems and processes used to maintain quality and compliance.
- Audit and inspection readiness: Conducting mock inspections and risk assessments to prepare a company for an official regulatory audit, such as an FDA inspection.
- Global market access: Providing guidance on how to meet the specific regulatory requirements of different countries, helping clients expand into new international markets.